Tumor
癌症序列变异解释和报告的标准和指南(ASCO和CAP联合推荐)
肺癌分析插件Can_28修复问题
Chom化疗插件及bug修复
Brca插件
FACTERA-fusionGene
SEGF-挖掘NGS中融合基因的新方法
maftools-肿瘤突变数据可视化神器
从数据库中获取免疫相关基因(IRGs)
数据库10KIP—基于ImmPort免疫组学数据挖掘
TCGA数据分析
下载分析TCGA数据库的数据
差异表达分析(limma & edgeR & DESeq2)
TCGA数据的规律【更新中】
生存分析
生存模型构建
突变数据
本文档使用 MrDoc 发布
-
+
up
down
首页
突变数据
首先是数据下载。 [TCGA突变数据的下载、整理和可视化](https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247487010&idx=1&sn=904fc43625d575f67f9f96ab3b04fc8b&scene=21#wechat_redirect) 突变数据在TCGA数据库中存储为maf格式,需要将其读入R语言,有很好的R包可以一键展示数据特征,形如: 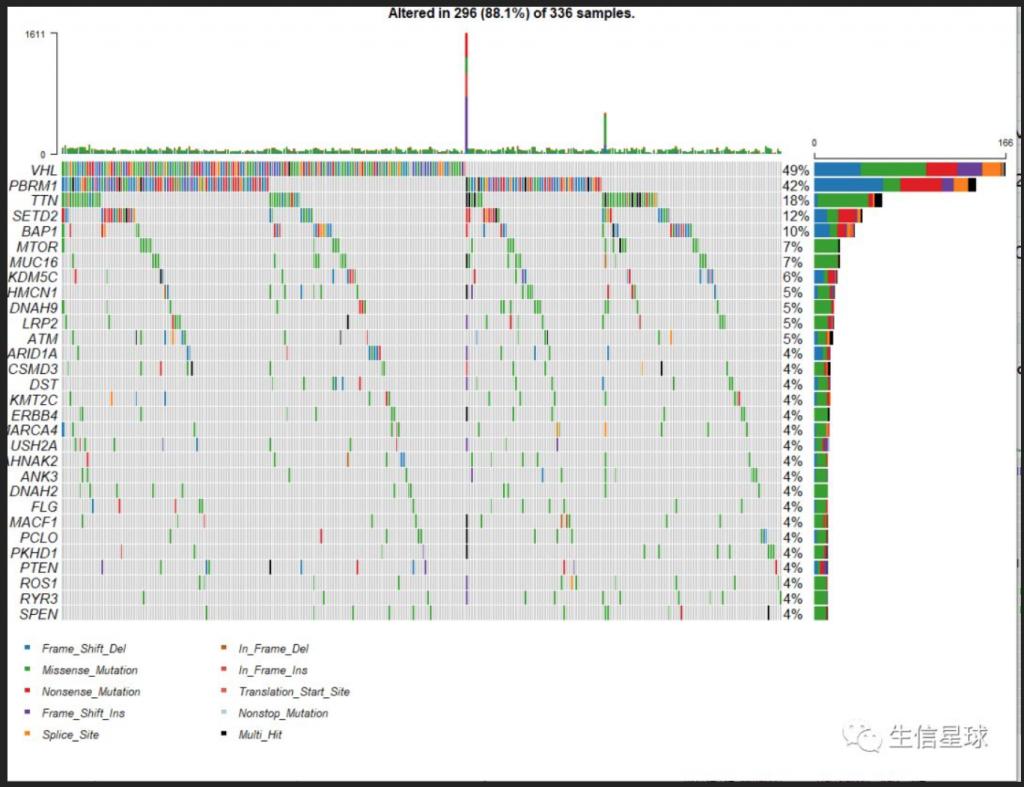 有哪些基因在较多样本中发生了突变,哪个样本突变的基因较多,所占的比例等等都可以一目了然。 还可以进行mutation signafiture分析 [TCGA突变数据分析-mutation signafiture](https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247487026&idx=1&sn=36f51f766aa846301902ff2b2069cab5&scene=21#wechat_redirect) 得到如下的图:  展示了30个signafiture 在所有样本中的分布。 我们还可以根据某个基因是否突变对样本进行分组,比较指定的基因在两组之间的表达量: 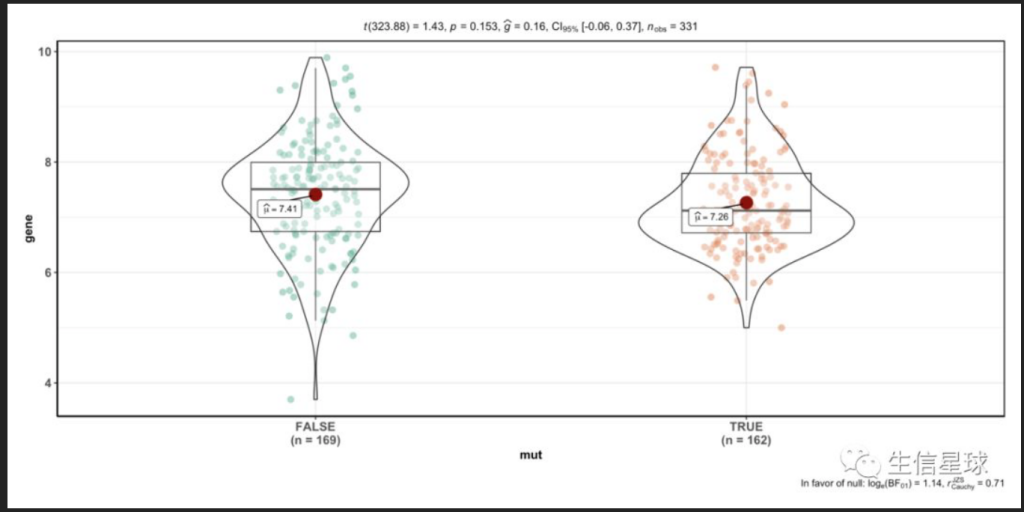 [任意基因的任意分组比较](https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247487032&idx=1&sn=83e7e9b21c124fdbfd3b3df25548be50&scene=21#wechat_redirect)(标题忘记改了) 也可以比较指定的两个基因在任意分组下的相关性: [表达矩阵任意两个基因的相关性分析和绘图](https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247487038&idx=1&sn=79a3f23e655fcde67523127ba26002b2&scene=21#wechat_redirect)  如果有幸发现两个基因本身表达量是相关的,但在根据突变分组后发现这种相关性被打破了,就有可能得到一个新故事,几率很小,可以用循环来筛选。 只要R语言基础牢固,想要根据什么分组都只是改改代码的问题,包括但不局限于是否突变、人种、性别等等。
laihui126
2023年1月10日 10:55
分享文档
收藏文档
上一篇
下一篇
微信扫一扫
复制链接
手机扫一扫进行分享
复制链接
关于 MrDoc
觅道文档MrDoc
是
州的先生
开发并开源的在线文档系统,其适合作为个人和小型团队的云笔记、文档和知识库管理工具。
如果觅道文档给你或你的团队带来了帮助,欢迎对作者进行一些打赏捐助,这将有力支持作者持续投入精力更新和维护觅道文档,感谢你的捐助!
>>>捐助鸣谢列表
微信
支付宝
QQ
PayPal
下载Markdown文件
分享
链接
类型
密码
更新密码



