Tumor
癌症序列变异解释和报告的标准和指南(ASCO和CAP联合推荐)
肺癌分析插件Can_28修复问题
Chom化疗插件及bug修复
Brca插件
FACTERA-fusionGene
SEGF-挖掘NGS中融合基因的新方法
maftools-肿瘤突变数据可视化神器
从数据库中获取免疫相关基因(IRGs)
数据库10KIP—基于ImmPort免疫组学数据挖掘
TCGA数据分析
下载分析TCGA数据库的数据
差异表达分析(limma & edgeR & DESeq2)
TCGA数据的规律【更新中】
生存分析
生存模型构建
突变数据
本文档使用 MrDoc 发布
-
+
up
down
首页
TCGA数据的规律【更新中】
长期积累TCGA数据中的规律。。。 ## TCGA条码(barcode)信息 [TCGA条码](https://docs.gdc.cancer.gov/Encyclopedia/pages/TCGA_Barcode/) 由一组标识符组成。每个都专门标识一个TCGA数据元素。有关元数据标识符如何组成条形码的说明,请参见下图: 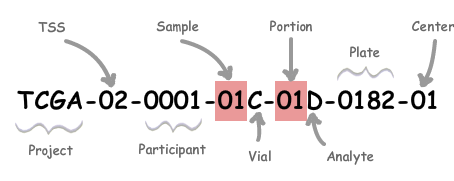 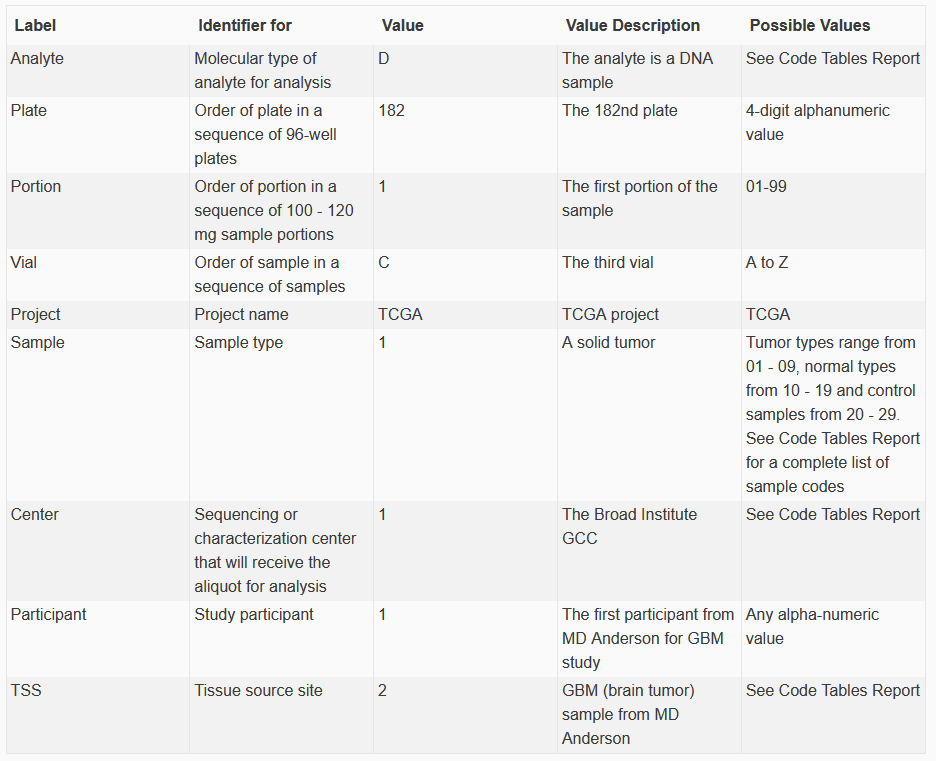 ### 用R语言解决一下TCGA的id中隐藏的分组信息  图中展示的是TCGA样本id,分组信息是在这个id的第14-15位,01-09是tumor,10-29是normal。我拿了一个示例数据,请在生信星球公众号回复0129获取。 根据这个生成一个分组信息它是一个向量形如  如果在id的14-15位在1-9之间就标记tumor,10-29之间就标记normal。这里面涉及到字符串截取、数据类型转换,%in%函数以及ifelse函数(if-else循环的变体) 将示例数据放到你的工作目录 ```R load(file="id.Rdata") table(substring(id,14,15)) #table看有多少重复值 num <- as.numeric(substring(id,14,15)) #截取出来是字符串,要转为数字 #屡试不爽的ifelse group_list=ifelse(num %in% 1:9,"Tumor","Normal") ``` 如果你要用自己的数据试试,就把样本编号命名为id,来跑一跑就ok。这一列信息后面作图会用。 ## RNA-Seq数据格式更新 > 2022年4月发现,TCGA的RNAseq数据悄悄更新了,选择一个project,例如TCGA-LUSC,公开数据,选择RNA-Seq后,workflow.type里只有"STAR- Counts"了,没有HTSeq-Counts了。   你会发现STAR-Counts里面有1106个文件,其中553个是Gene Expression Quantification(.rna_seq.augmented_star_gene_counts.tsv),这是我们合并表达谱所需要的文件。另外553个文件是Splice Junction Quantification(*.rna_seq.star_splice_junctions.tsv.gz),这个主要是检测新的转录本或者融合的文件,且他们都是无权限查看的(controlled)。  下载着553个Gene Expression Quantification文件: ``` query_TCGA = GDCquery(project="TCGA-LUSC", data.category="Transcriptome Profiling", experimental.strategy="RNA-Seq", workflow.type="STAR - Counts", access="open") GDCdownload(query = query_TCGA, files.per.chunk = 10) ``` ## STAR-counts的文件说明 每个文件夹里面会有一个star_gene_counts.tsv,我们可以随便打开一个看一下,这个文件的内容跟老版本的完全不一样,包含的信息更多。甚至包含了RNA类型,这样就能很容易的区分mRNA和lncRNA了,另外还包含的基因名和ID。 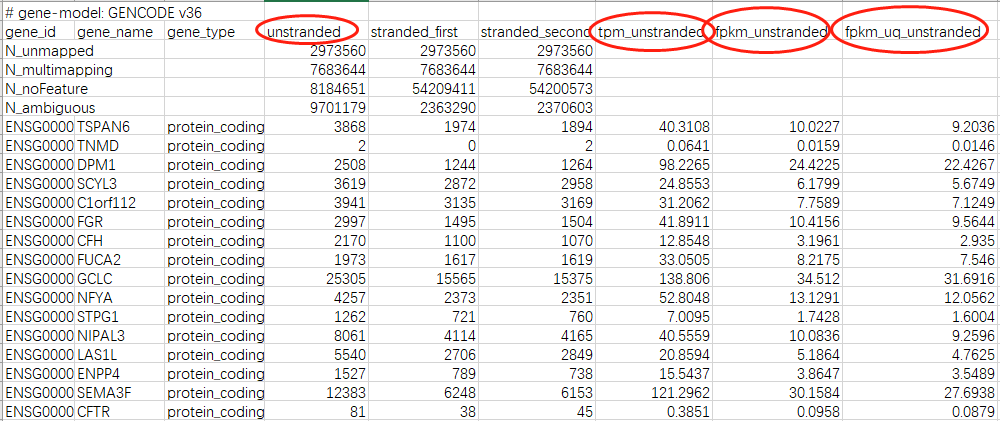 文件中,除了有STAR-counts,还有TPM,FPKM和FPKM_UQ。这几个数据的具体计算方法可以参考 [TCGA官方文档](https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/) > STAR-counts的计算比较简单,表示有几条reads比对到相应的基因上面:  > dFPKM The fragments per kilobase of transcript per million mapped reads (FPKM) calculation aims to control for transcript length and overall sequencing quantity. > wFPKM-UQ(Upper Quartile FPKM) The upper quartile FPKM (FPKM-UQ) is a modified FPKM calculation in which the protein coding gene in the 75th percentile position is substituted for the sequencing quantity. This is thought to provide a more stable value than including the noisier genes at the extremes. > iTPM The transcripts per million calculation is similar to FPKM, but the difference is that all transcripts are normalized for length first. Then, instead of using the total overall read count as a normalization for size, the sum of the length-normalized transcript values are used as an indicator of size.  示例解释: ``` Examples Sample 1: Gene A Gene length: 3,000 bp 1,000 reads mapped to Gene A 1,000,000 reads mapped to all protein-coding regions Read count in Sample 1 for 75th percentile gene: 2,000 Number of protein coding genes on autosomes: 19,029 Sum of length-normalized transcript counts: 9,000,000 FPKM for Gene A = 1,000 * 10^9 / (3,000 * 50,000,000) = 6.67 FPKM-UQ for Gene A = 1,000) * 10^9 / (3,000 * 2,000 * 19,029) = 8.76 TPM for Gene A = (1,000 * 1,000 / 3,000) * 1,000,000 / (9,000,000) = 37.04 ```
laihui126
2023年1月12日 14:57
分享文档
收藏文档
上一篇
下一篇
微信扫一扫
复制链接
手机扫一扫进行分享
复制链接
关于 MrDoc
觅道文档MrDoc
是
州的先生
开发并开源的在线文档系统,其适合作为个人和小型团队的云笔记、文档和知识库管理工具。
如果觅道文档给你或你的团队带来了帮助,欢迎对作者进行一些打赏捐助,这将有力支持作者持续投入精力更新和维护觅道文档,感谢你的捐助!
>>>捐助鸣谢列表
微信
支付宝
QQ
PayPal
下载Markdown文件
分享
链接
类型
密码
更新密码



