Tumor
癌症序列变异解释和报告的标准和指南(ASCO和CAP联合推荐)
肺癌分析插件Can_28修复问题
Chom化疗插件及bug修复
Brca插件
FACTERA-fusionGene
SEGF-挖掘NGS中融合基因的新方法
maftools-肿瘤突变数据可视化神器
从数据库中获取免疫相关基因(IRGs)
数据库10KIP—基于ImmPort免疫组学数据挖掘
TCGA数据分析
下载分析TCGA数据库的数据
差异表达分析(limma & edgeR & DESeq2)
TCGA数据的规律【更新中】
生存分析
生存模型构建
突变数据
本文档使用 MrDoc 发布
-
+
up
down
首页
下载分析TCGA数据库的数据
# 一、如何下载TCGA数据库的数据? 1、打开网页https://cancergenome.nih.gov/  点击进入“Launch Data Portal”, 即GDC数据库的界面,可以看到分类很详细,  2、以下载open的MAF文件进行示例。按上图,点击“Repository”,进入数据下载界面。 maf格式的mutation记录文件在TCGA里面已经是level4的数据啦,所以是完全open的,可以随意下载,只需要去其GDC官网简单点击,选择即可。主要步骤就是在https://portal.gdc.cancer.gov/repository 里面点击过滤文件类型,选择maf格式,再过滤access权限,选择open即可,最后得到的132个文件就是我们需要的。 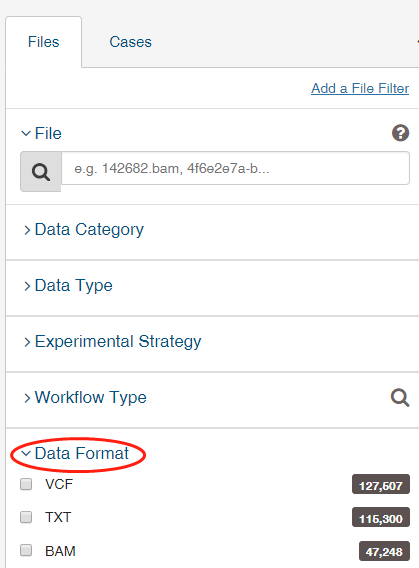 在数据格式这儿选择MAF格式,看到共有318个。右边展示的数据为controlled表示未开放,下载不了的,需要获得权限才可下载。  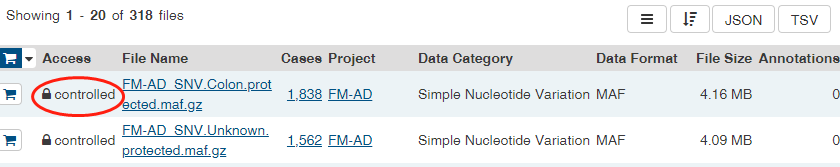 而上面有一个汇总展示图,先点击“Show More”,展开后看得到还有“Access Level”,选择“OPEN”,看到共有132个是可下载的。 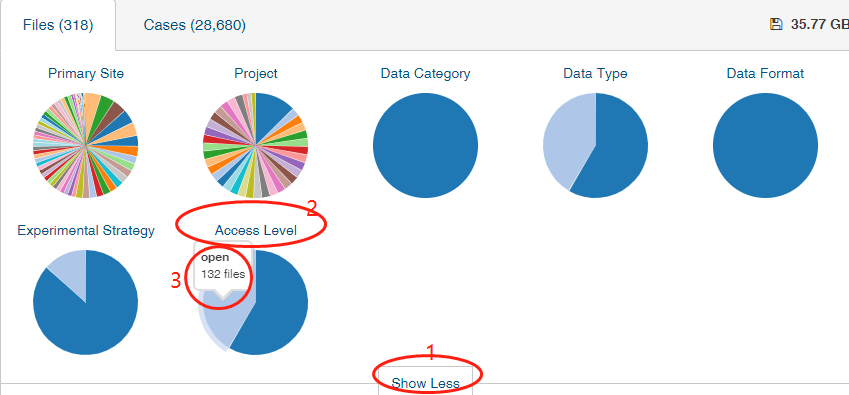 点击购物车按钮(Cart),Add all files to the Cart. 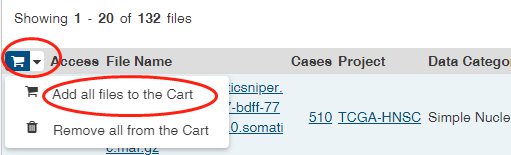 3、下载Manifest文件。从主选项卡那儿进入Cart,可以看到有132个文件总大小为2.5G多。点击“Download”,选择下载Manifest文件,获得了文件gdc_manifest_20181123_005852.txt,用于后续下载数据。 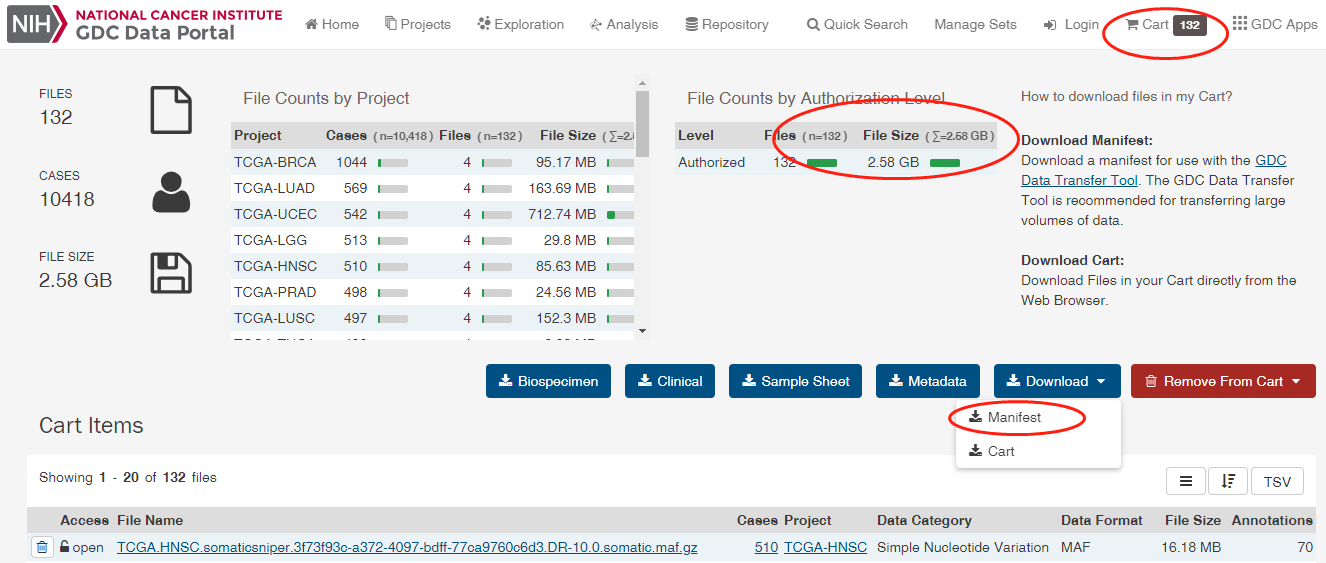 4、通过GDC工具进行下载。 (1)该工具可以在选项卡上的“GDC Apps”中下载,即“Data Transfer Tool”,该工具有windows、linux等版本。我们以linux为例。 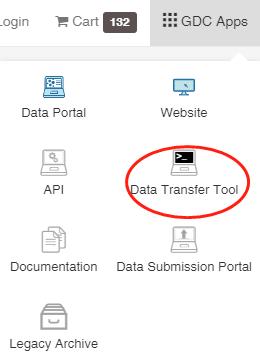 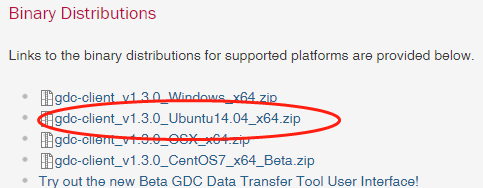 (2)下载编译后的二进制版本,可直接进行使用。 使用方法可以具体看GDC使用方法:http://docs.gdc.cancer.gov/Data_Transfer_Tool/Users_Guide/Getting_Started/ (3)下载主要分两步:  第一步,即下载Manifest文件或者UUID(针对获得了controlled文件权限)。 第二步,下载或者Upload。 (4)Download的基本命令: ``` #help帮助 gdc-client --help #download help gdc-client download --help #download准备好的132个MAF文件的Manifest文件 gdc-client download -m gdc_manifest_20181123_005852.txt #即可下载, -d 指定下载到目标目录 ``` 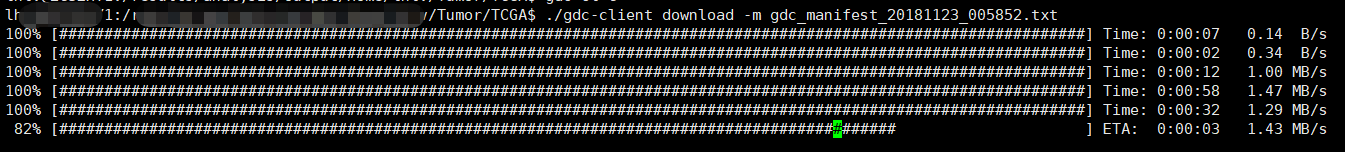 看到目录中已经有了*.maf.gz文件,而该文件的前面是Project号(TCGA)和癌种(HNSC),以及变异检测软件(Mutect)。 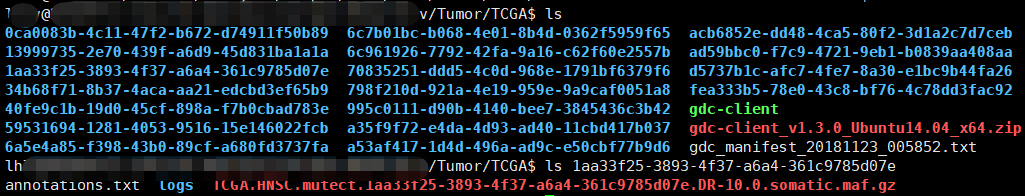 # 二、以TCGA的数据分析panel的TMB 1、下载569个LUAD的体细胞突变的maf文件(示例) 首先,选好的条件是这样的:TCGA-LUAD && open && MAF 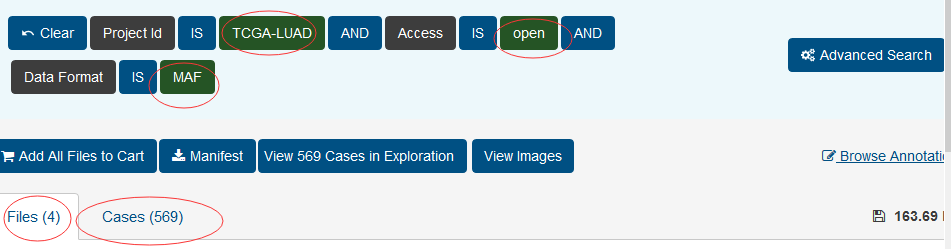 可以看到总共有569例Cases,但是显示文件只有4个。他们分别是四种软件call的变异集合(mutect, somaticsniper,varscan,muse)。 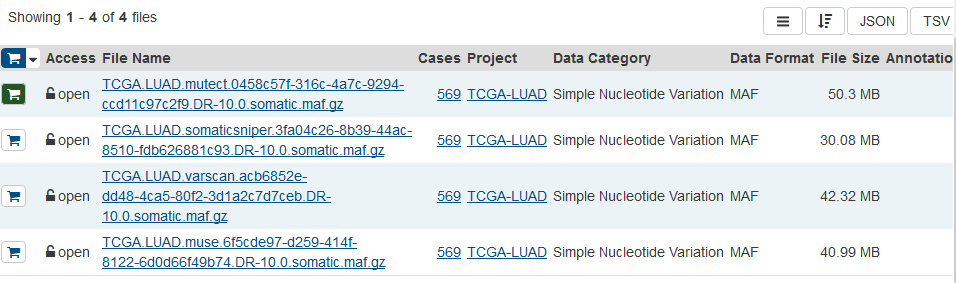 其次,我们将所有假如到Cart。然后下载Manifest文件,用于gdc下载。 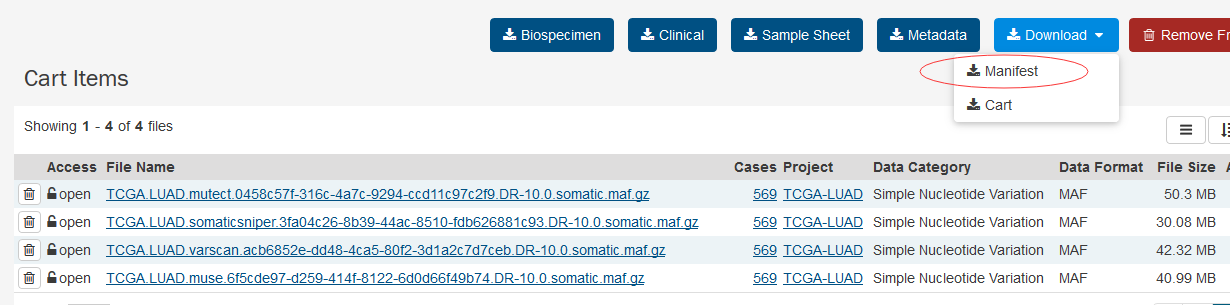 最后,进行gdc下载: `../gdc-client download -m gdc_manifest_20181214_011958.txt #P30`  2、对于TCGA的maf文件格式,可以参照:https://docs.gdc.cancer.gov/Data/File_Formats/MAF_Format/ 上面的介绍。 ## 数据下载方法 有三种下载方式,他们的目的是一样的,就是整理出表达矩阵和临床信息两个表格,使用体验各有不同。 形如: ``` expr[1:4,1:4] #> TCGA-A3-3307-01A-01T-0860-13 TCGA-A3-3308-01A-02R-1324-13 #> hsa-let-7a-1 5056 14503 #> hsa-let-7a-2 10323 29238 #> hsa-let-7a-3 5429 14738 #> hsa-let-7b 17908 37062 #> TCGA-A3-3311-01A-02R-1324-13 TCGA-A3-3313-01A-02R-1324-13 #> hsa-let-7a-1 8147 7138 #> hsa-let-7a-2 16325 14356 #> hsa-let-7a-3 8249 7002 #> hsa-let-7b 28984 6909 clinical[1:4,1:4] #> patient.vital_status patient.days_to_death #> TCGA-3Z-A93Z alive <NA> #> TCGA-6D-AA2E alive <NA> #> TCGA-A3-3306 alive <NA> #> TCGA-A3-3307 alive <NA> #> patient.days_to_last_followup patient.race #> TCGA-3Z-A93Z 4 black or african american #> TCGA-6D-AA2E 135 black or african american #> TCGA-A3-3306 1120 white #> TCGA-A3-3307 1436 <NA> ``` ## 1. 官方工具GDC 需要去官网下载对应系统版本的GDC软件,存放在工作目录下。 关于这个工具前后写了三篇: (1)[GDC数据下载](https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247486909&idx=1&sn=a4bc91129be472edb6e470bffd92b129&scene=21#wechat_redirect) (2)[GDC数据整理](https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247486924&idx=1&sn=70cf1a4428b837d26495d0ac1fc5d6f9&scene=21#wechat_redirect) (3)[GDC数据整理续集](https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247486988&idx=1&sn=42323e2b8796a465edfbe5f7a9dade10&scene=21#wechat_redirect) 这个方法需要扎实的的linux命令行和R语言基础,仅仅是理解代码,也是需要花费一些时间的。 ## 2.R包TCGAbiolinks下载数据 R包TCGAbiolinks下载数据,这是一个完全基于R语言的流程,下载的是最新的数据。 ```R if(!requireNamespace("BiocManager", quietly = TRUE)){ install.packages("BiocManager") } BiocManager::install("TCGAbiolinks") ``` ### 下载方法 该包可以使用2种方法下载GDC数据,分别是: - client: 此方法创建MANIFEST文件并使用 GDC Data Transfer Tool下载数据。此方法更可靠,但与api方法相比可能更慢。 - api: 此方法使用 GDC Application Programming Interface (API)下载数据。这将创建一个MANIFEST文件,并且下载的数据将是一个格式为tar.gz的压缩文件。文件过大或数量过多可能会导致下载失败,可以使用参数files.per.chunk将文件拆分成多个小文件下,例如,chunks.per.download=5,则将每个tar.gz分为5个小文件下载。 ### GDCquery()参数解析 (1)project: 可以使用`TCGAbiolinks:::getGDCprojects()$project_id`得到各个癌种的项目id,总共有74个ID值。  (2)data.category: 总共有11种。 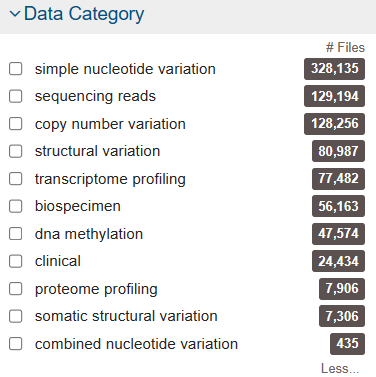 可使用`TCGAbiolinks:::getProjectSummary(project)`查看project中有哪些数据类型,如查询"TCGA-BRCA",有9种数据类型(和网页GDC查询到的是一样的),case_count为病人数,file_count为对应的文件数。要下载表达谱,可以设置data.category="Transcriptome Profiling"。 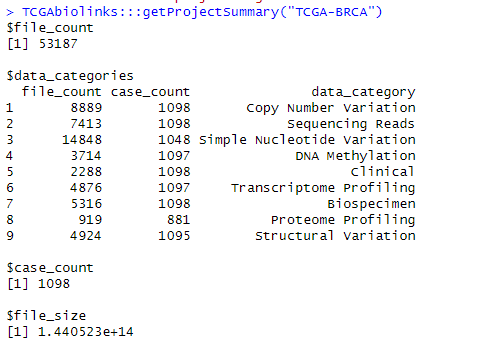 (3)data.type:徐泽下载文件的数据类型。没有命令可以查看data.type里都有哪些数据类型,通过官网查看data.type,目前已有28种,但设置参数的时候不代表所有的project和data.category都对应28种。需要先在官网查看,再设置。 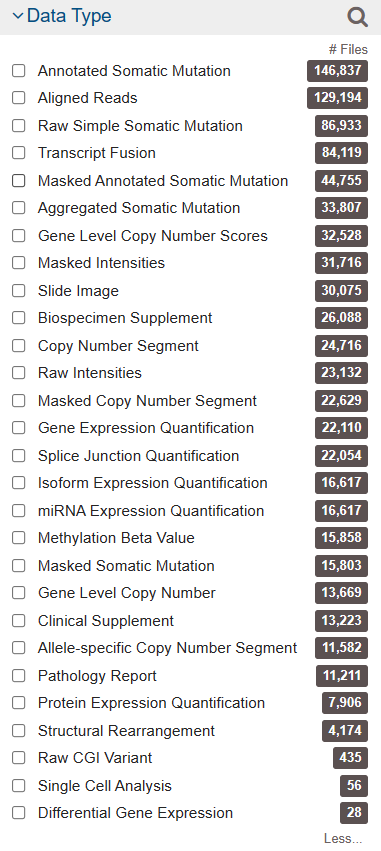 例如, ``` #下载RNAseq的counts数据 data.type ="Gene Expression Quantification" #下载miRNA数据 data.type ="miRNA Expression Quantification" ``` (4)workflow.type:该数据类型有很多种(目前39种),依data.type选择而不同。比如 Gene Expression Quantification 数据类型下workflow.type有3种类型分别为: - STAR - Counts - CellRanger - 10x Filtered Counts - CellRanger - 10x Raw Counts 具体可在GDC官网查看 (5)legacy: 这个参数主要是因为TCGA数据有两个入口可以下载,GDC Legacy Archive 和 GDC Data Portal。 | | Legacy Archive | Data Portal| | --- | --- | --- | | 注释基因组版本 | hg19 | hg38 | | 建议 | DNA层面数据 | 转录组数据 | > 该参数默认是hg38的GDC Data Portal, 点做SNP分析的时候很多数据库没有hg38版本的数据,都是hg19的。 (6)access:数据开放和不开放,有两个参数:controlled, open。 (7)platform:涉及到的平台种类很多,具体官网看每种数据都有什么平台的可以下载。该参数可省略不设。 (8)file.type: 主要是在GDC Legacy Archive下载数据的时候使用,可以参考官网说明。在GDC Data Portal下载数据,该参数省略不设置。 (9)barcode:A list of barcodes to filter the files to download。可以根据这个参数设置只下载某个样本等。如: `barcode = c("TCGA-14-0736-02A-01R-2005-01","TCGA-06-0211-02A-02R-2005-01")` (10)experimental.strategy: 两个下载入口参数选择: - GDC Data Portal:WXS, RNA-Seq, miRNA-Seq, Genotyping Array - Legacy: WXS, RNA-Seq, miRNA-Seq, Genotyping Array, DNA-Seq, Methylation array, Protein expression array, WXS,CGH array, VALIDATION, Gene expression array,WGS, MSI-Mono-Dinucleotide Assay, miRNA expression array, Mixed strategies, AMPLICON, Exon array, Total RNA-Seq, Capillary sequencing, Bisulfite-Seq (11)sample.type: A sample type to filter the files to download,可以对样本类型进行过滤下载。这里我要下载所有样本类型数据,不设置。部分值选择如下(全部可以查看官网):如sample.type = "Recurrent Solid Tumor" > 其他参数可以看 [文档](http://www.bioconductor.org/packages/release/bioc/manuals/TCGAbiolinks/man/TCGAbiolinks.pdf) ### GDCdownload参数介绍 上面介绍了GDCquery这个函数,他下载数据时要用到的函数,除此以外,我们还需要GDCdownload函数。GDCdownload函数使用GDC API或GDC传输工具下载GDC数据,用户可以使用查询参数查询的数据将保存在一个文件夹中:project/data.category。函数的整体框架为: `GDCdownload(query, token.file, method ="api", directory ="GDCdata",files.per.chunk =NULL)` (1) query:这个参数就是来自GDCquery的结果。 (3) method:使用API (POST方法)或gdc客户端工具。选择“api”,“client”。API更快,但是下载过程中数据可能会损坏,可能需要重新执行。 (2) token.file:这个是下载受限的文件(仅适用于method=“client”),一般下载用不到。 (4) directory:下载数据的存放目录/文件夹。默认:GDCdata。 (5) files.per.chunk:这将使API方法一次只下载n个(files.per.chunk)文件。当数据量过大时,可能会下载出错,可设置files.per.chunk参数减少下载出错问题。值为整数,即可将文件拆分为几个文件下载,如files.per.chunk = 6。 ### 临床信息下载 ``` # 方法1 clinical = GDCquery_clinic(project = "TCGA-LUSC", type = "clinical") #共有 70个变量,504个cases # 写到cssv文件 write.csv(file="clinical.csv", clinical) # 方法2 query_clinical = GDCquery(project ="TCGA-LUSC", data.category ="Clinical") #然后在使用 GDCdownload(query_clinical, files.per.chunk = 20)进行下载 ``` ``` # 查看信息 dim(clinical) [1] 504 70 #共有504个样本,70条临床信息 names(clinical) #查看具体信息 ``` ### 下载案例 ```R query <- GDCquery(project ="TCGA-ACC",data.category ="Copy number variation", legacy = TRUE, file.type ="hg19.seg", barcode = c("TCGA-OR-A5LR-01A-11D-A29H-01","TCGA-OR-A5LJ-10A-01D-A29K-01")) # 数据将被保存在 GDCdata/TCGA-ACC/legacy/Copy_number_variation/Copy_number_segmentation GDCdownload(query, method ="api")## Not run: # 从XML下载临床数据 query <- GDCquery(project ="TCGA-COAD", data.category ="Clinical") GDCdownload(query, files.per.chunk = 200) query <- GDCquery(project ="TARGET-AML",data.category ="Transcriptome Profiling",data.type ="miRNA Expression Quantification",workflow.type ="BCGSC miRNA Profiling",barcode = c("TARGET-20-PARUDL-03A-01R","TARGET-20-PASRRB-03A-01R"))# 数据将被保存在:# example_data_dir/TARGET-AML/harmonized/Transcriptome_Profiling/miRNA_Expression_Quantification GDCdownload(query, method ="client", directory ="example_data_dir") ``` 应用案例(下载TCGA-LUSC数据): ```R # query query_TCGA = GDCquery(project="TCGA-LUSC", data.category="Transcriptome Profiling", experimental.strategy="RNA-Seq", workflow.type="STAR - Counts", access="open") lusc_res = getResults(query_TCGA) #查看query结果 dim(lusc_res) colnames(lusc_res) head(lusc_res) # download data (总文件较大时,files.per.chunk需要设置小一些) GDCdownload(query = query_TCGA, files.per.chunk = 10) # 数据保存在目录:GDCdata\TCGA-LUSC\harmonized\Transcriptome_Profiling\Gene_Expression_Quantification ``` ## 3.R包RTCGA [使用RTCGA包获取数据](https://mp.weixin.qq.com/s?__biz=MzU4NjU4ODQ2MQ==&mid=2247486960&idx=1&sn=16e2db5c1d9a98a86f7e7fe18be27845&scene=21#wechat_redirect) 这是一个数据库式的包,把所有数据都包装进去了,导致包很大,不是最新的数据,但最简单。 总结一下这三种方法,都是分别下载表达矩阵和meta信息,但由于有的病人既有肿瘤样本,又有正常样本,导致他们并非是一一对应的关系,需要一定的R语言技巧。
laihui126
2023年1月12日 15:25
分享文档
收藏文档
上一篇
下一篇
微信扫一扫
复制链接
手机扫一扫进行分享
复制链接
关于 MrDoc
觅道文档MrDoc
是
州的先生
开发并开源的在线文档系统,其适合作为个人和小型团队的云笔记、文档和知识库管理工具。
如果觅道文档给你或你的团队带来了帮助,欢迎对作者进行一些打赏捐助,这将有力支持作者持续投入精力更新和维护觅道文档,感谢你的捐助!
>>>捐助鸣谢列表
微信
支付宝
QQ
PayPal
下载Markdown文件
分享
链接
类型
密码
更新密码



