Tumor
癌症序列变异解释和报告的标准和指南(ASCO和CAP联合推荐)
肺癌分析插件Can_28修复问题
Chom化疗插件及bug修复
Brca插件
FACTERA-fusionGene
SEGF-挖掘NGS中融合基因的新方法
maftools-肿瘤突变数据可视化神器
从数据库中获取免疫相关基因(IRGs)
数据库10KIP—基于ImmPort免疫组学数据挖掘
TCGA数据分析
下载分析TCGA数据库的数据
差异表达分析(limma & edgeR & DESeq2)
TCGA数据的规律【更新中】
生存分析
生存模型构建
突变数据
本文档使用 MrDoc 发布
-
+
up
down
首页
SEGF-挖掘NGS中融合基因的新方法
# SEGF思路: 1、通过Target_reference_prepare.pl和bed文件提取fasta中对应的target范围; `perl $Bin/script/Target_reference_prepare.pl $Bin/database/by_chr/$tmp[0].fa $tmp[1] $len $tmp[3] ./reference/reference.fa` ``` >ALK CTTAGTCATTACAAATAACTCCTTTATTTCCGTTCCCTCTCCCCTCAAAT GGCTCATGTCCACATCAACAAGGCAAGGAAACATCTATGACCCCAACTAT ... >EML4 cggcgcggcgcTCGCGGCTGCTGCCTGGGAGGGAGGCCGGGCAGGCGGCT GAGCGGCGCGGCTCTCAACGTGACGGGGAAGTGGTTCGGGCGGCCGCGGC ``` 并对reference.fa建库 `$Bin/bin/formatdb -i ./reference/reference.fa -p F -o T` 2、对下机数据fq.gz文件进行处理。Deal_reads.pl脚本,-trim_len设置5'和3‘端切除碱基数(一般reads两端质量低),-remain_len设置保留reads两端的长度。生成用于blast和soap比对的fasta文件。 `perl $Bin/script/Deal_reads.pl $fq $trim_len $remain_len ./deal_fastq/$basename.blast.fa ./deal_fastq/$basename.soap.fa` 如下,可以看到reads名称的对应关系: ==> deal_fastq/s2_R1.fastq.gz.blast.fa <== ``` >NB501918:39:HVHH5AFXX:1:11101:7916:1025/1 AGTCTTCTTGGCTTCCTTCCTTTTCCCTAAGGTTGTGTCAATATTTTGGATTTTTATAGTCATTTCCCTTTCTCTGTGGT >NB501918:39:HVHH5AFXX:1:11101:25696:1027/1 CCATCATCCTGACGTTGGGTCGGCTGATACACCCCTGTCCTCATCCAGCTGGTGGATGTGTTATACAACGAAGAGAAGCA ==> deal_fastq/s2_R1.fastq.gz.soap.fa <== >NB501918:39:HVHH5AFXX:1:11101:7916:1025/1-1/1 AGTCTTCTTGGCTTCCTTCCTTTTCCCTAAGGTTGTGTCA >NB501918:39:HVHH5AFXX:1:11101:7916:1025/1-1/2 ATATTTTGGATTTTTATAGTCATTTCCCTTTCTCTGTGGT >NB501918:39:HVHH5AFXX:1:11101:25696:1027/1-1/1 CCATCATCCTGACGTTGGGTCGGCTGATACACCCCTGTCC >NB501918:39:HVHH5AFXX:1:11101:25696:1027/1-1/2 TCATCCAGCTGGTGGATGTGTTATACAACGAAGAGAAGCA ``` 3、进行blast和soap2分别比对(均为SE比对)。 ``` #blastall $Bin/bin/blastall -p blastn -i ./deal_fastq/$basename.blast.fa -d ./reference/reference.fa -o ./tmp/$basename.blast -e 1e-10 -F F -m 8 -a $process #soap2.21 $Bin/bin/soap2.21 -a ./deal_fastq/$basename.soap.fa -D $Bin/database/hg19.fasta.index -o ./tmp/$basename.soap -p $process -s $remain_len ``` 分别得到结果如下: ``` >head tmp/s2_R1.fastq.gz.blast NB501918:39:HVHH5AFXX:1:11101:10545:1045/1 ALK 100.00 42 0 0 1 42 320765 320806 4e-17 83.8 NB501918:39:HVHH5AFXX:1:11101:10545:1045/1 ALK 97.67 43 1 0 1 43 480530 480488 2e-15 77.8 >head tmp/s2_R1.fastq.gz.soap NB501918:39:HVHH5AFXX:1:11101:7916:1025/1-1/1 AGTCTTCTTGGCTTCCTTCCTTTTCCCTAAGGTTGTGTCA hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh 1 a 40 + chr7 99944690 0 40M 40 NB501918:39:HVHH5AFXX:1:11101:7916:1025/1-1/2 ATATTTTGGATTTTTATAGTCATTTCCCTTTCTCTGTGGT hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh 1 a 40 + chr7 99944780 0 40M 40 NB501918:39:HVHH5AFXX:1:11101:1258:1039/1-1/1 GAACTAGGGAGAGGCTGCCTTTTCATGGGAAAAAGACATT hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh 1 a 40 - chr9 116745903 0 40M 40 NB501918:39:HVHH5AFXX:1:11101:1258:1039/1-1/2 ACTCAGGCAGGAATCTGGCTGGTCACGCAGCTGCAGCTGT hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh 1 a 40 - chr9 116745813 1 T->6G40 40M 6T33 ``` 4、对结果进行处理Result-filter.pl `perl $Bin/script/Result-filter.pl ./tmp/$basename.blast ./tmp/$basename.soap ./Result/$basename.fusions.txt` 这儿需要熟悉blast和soap2比对结果。soap2比对结果解释如下: ``` 从左到右 1. 编号: read 的编号 2. read的序列.如果read比对上参考序列的负链,会被反向互补为正链 3. 质量值:序列的质量值,和序列顺序一致,如果read反向互补,质量值也会随着改变 4. 比对上的次数: 最优比对的次数。没有比对上的read将被忽略 5. a/b:pair-end比对的标记, 表示read属于来自哪个文件 6. 长度: read长度,如果是容缺失的比对,长度将是加上缺失片断的长度 7. +/-: 比对上参考序列的正链或负链 8. 染色体名称:参考序列的染色体名称 9. 位点:第一个碱基在染色体上的位置,从1开始 10. 错配的个数 11. 错配的详细信息("A->11T16" 意思是一个错配,在参考序列的位置是第11个(从0开始),在参考序列上是A,rea上是T,质量值是16) 12. 比对上的数目 ("44M" 意思是44个碱基比对上了) 13. 对比的细节 ("11A32"意思是前10个比对上了,第11个是错配(从0开始),后面32个还是比对上了) ``` (1)该程序中使用了soap2的第1、2、4、10列信息,即read名称、read序列、最优比对次数、错配碱基数。由于先前构建用于soap2比对的fasta文件是将每条read两端拆出特定长度作为read1(-1/1或-2/1)和read2(-1/2或-2/2),因此首先需要两条read1和read2名称一致(即同一序列),且这两条短reads的最优比对次数均为1,错配碱基数均为0. 以此read名建立哈希。 blast结果总共12列:Query id, Subject id, % identity, alignment length, mismatches, gap openings, q. start, q. end, s. start, s. end, e-value, bit score (2)判断blast结果的read名称在上面建立的哈希中存在,且100%比对,reads比对上的长度为拆的时候设置的特定长度(可更该为大于等于改长度),错配碱基数为0,gap数为0;read比对上的起始和终止为1和35或36和70(和设定特定长度有关,可更改),且相同read名称的下一个比对结果,同样符合这些条件,且上下两个比对上的read的起始位置不一样,终止位子也不一样,第二列的比对上的reference也不一样。 5、获得结果文件 ``` ID1 Sequence ID2 Gene1 Gene2 Query1_Start Query1_End Query2_Start Query2_End Ref1_Start Ref1_End Ref2_Start Ref2_End NB501918:39:HVHH5AFXX:2:11107:2825:17780/1 TAGGTGGCCTTTTTATTGTTGATTGTTTCCTGGCA<->GTGGGAGCTAGAAGTGACGTCTAGGGGTGGGGGCG NB501918:39:HVHH5AFXX:2:11107:2825:17780/1 EML4 ALK 1 35 36 70 110704 110738 32391 32357 NB501918:39:HVHH5AFXX:2:11107:2825:17780/1 TAGGTGGCCTTTTTATTGTTGATTGTTTCCTGGCA<->GTGGGAGCTAGAAGTGACGTCTAGGGGTGGGGGCG NB501918:39:HVHH5AFXX:2:11107:2825:17780/1 ALK EML4 36 70 1 35 32391 32357 110704 110738 NB501918:39:HVHH5AFXX:2:11107:2825:17780/1 TAGGTGGCCTTTTTATTGTTGATTGTTTCCTGGCA<->GTGGGAGCTAGAAGTGACGTCTAGGGGTGGGGGCG NB501918:39:HVHH5AFXX:2:11107:2825:17780/1 EML4 ALK 1 35 36 70 110704 110738 32391 32357 NB501918:39:HVHH5AFXX:2:11107:2825:17780/1 TAGGTGGCCTTTTTATTGTTGATTGTTTCCTGGCA<->GTGGGAGCTAGAAGTGACGTCTAGGGGTGGGGGCG NB501918:39:HVHH5AFXX:2:11107:2825:17780/1 ALK EML4 36 70 1 35 32391 32357 110704 110738 NB501918:39:HVHH5AFXX:3:11409:22663:7783/1 TAGGTGGCCTTTTTATTGTTGATTGTTTCCTGGCA<->GTGGGAGCTAGAAGTGACGTCTAGGGGTGGGGGCG NB501918:39:HVHH5AFXX:3:11409:22663:7783/1 EML4 ALK 1 35 36 70 110704 110738 32391 32357 NB501918:39:HVHH5AFXX:3:11409:22663:7783/1 TAGGTGGCCTTTTTATTGTTGATTGTTTCCTGGCA<->GTGGGAGCTAGAAGTGACGTCTAGGGGTGGGGGCG NB501918:39:HVHH5AFXX:3:11409:22663:7783/1 ALK EML4 36 70 1 35 32391 32357 110704 110738 ``` # SEGF-挖掘临床二代数据中融合基因的新方法【转载】 元码又一专利已经进入实质审查阶段,这个专利就是基因融合分析新方法—SEGF (Single-end Gene Fusion),相关文章也已于 7 月份发表在《Genes》杂志上。今天小编将为您简单介绍一下这一方法。 首先,什么是基因融合? 基因融合是由于染色体断裂和重排导致的两个基因融合在一起的现象,是结构变异的一种,在肿瘤发生中起着关键作用。许多基因融合被认为是诊断标记,例如,9 号和 22 号染色体的易位现象导致 BCR 和 ABL1 基因之间产生了融合,可作为慢性粒细胞白血病(CML)的诊断标记,该易位现象导致生成费城染色体,在 CML 发生率高达 90%[1]。 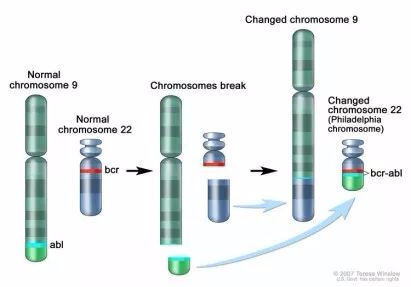 图 1 BCR 与 ABL1 融合产生费城染色体 [2] 随着下一代测序 (NGS) 和目标区域捕获技术的发展与应用,对从高通量数据中精确分析出融合基因方法的需求越来越大。 那么目前的分析方法有哪些呢?SEGF 又是怎么个优秀法呢? 目前分析结构变异的方法主要包括以下四种: 1. De novo assembly:该方法是检测结构变异的最直接的方法,且不受 PCR 结果的影响,但是由于基因组中重复区域的影响,我们很难将 NGS 的短序列正确地组合起来。而且,全基因组测序价格昂贵,不是融合基因检测的最优选择。  图 2 De novo assembly 方法检测结构变异原理图 [3] 2. Read-depth:该方法假设比对到基因组上的序列深度符合泊松分布,然后用实际的序列深度分布来判断结构变异。Read-depth 方法适合检测缺失和重复区,缺点是受实验环节的扩增和测序偏向性影响较大,结果往往不是很准确。 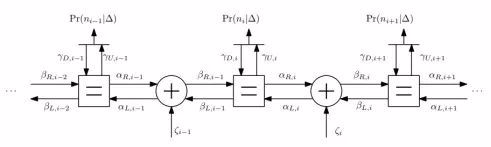 图 3 基于 Read-depth 方法利用隐马尔可夫模型检测结构变异原理 3. Read-pair:该方法利用 pair-end 的一对 reads 的中间空缺长度和序列方向特征进行检测。这种方法可以检测到大片段的插入、缺失、倒位、易位等结构变异,但受限于测序文库片段长度的偏差,同时,软件的参数设置对结果的影响也非常大。 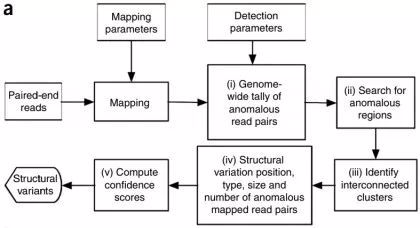 图 4 Read-pair 方法检测结构变异原理图 [5] 4. Split-read:该方法是分割短序列的方法。当基因组发生某一段的缺失,或转录组的剪接后,在测序过程中,横跨缺失位点及剪接位点的 reads 比对到基因组时,一条 reads 被切成两段,匹配到不同的区域,这样的序列叫做 soft-clipped reads,这些 reads 对于鉴定染色体结构变异及外源序列整合具有重要作用,soft-clip 的位置往往包含结构性变异的断点之处。该方法主要用于检测缺失、插入、倒置、易位等,但是基因组上的重复序列对结果影响较大,且软件的参数设置需要慎重考虑。 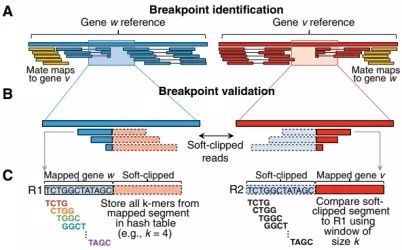 图 5 Split-read 方法检测结构变异原理图 [6] To be, or not to be, that is the question. 鉴于以上方法的优缺点,不屈的元码人选择另辟蹊径,启发于 split-read 方法,从单端 DNA-Seq 数据出发,开发了 SEGF,一种全新的融合基因分析方法。该方法基于 Linux 平台使用 PERL 语言开发,以压缩形式的原始测序数据 (FASTQ.gz) 为输入,去除两端质量低的部分序列后,对两端高质量的 35bp (默认) 使用常用的比对方法 BLAST 和 SOAP 进行比对,结合严格的过滤参数来获得融合候选序列,并进行基因区间的注释。 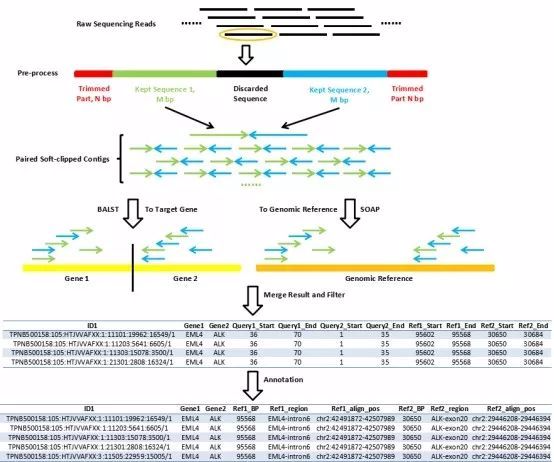 图 6 SEGF 检测基因融合原理图 为了评估 SEGF 方法的可靠性,我们将两种比对方法和两种结构变异分析方法相结合,与 SEGF 进行比较。其中 BWA 是比对软件,其优势是速度快且准确度高,它有 ALN 和 MEM 两种比对方法,分别应用了全局比对和局部比对的算法、针对于短序列和长序列。FACTERA 和 CREST 则是业内结构变异分析的两个知名软件 [6-7]。 在利用五种方法分析来自标准品 (n = 22/23) 和临床样本 (n = 11/286) 的融合基因时,SEGF 展现了最高的灵敏度和准确性。而且与其他方法相比,SEGF 能够通过单端 NGS 测序数据检测复杂的基因融合类型,节省了一半的计算时间。 表 1 应用于标准品上的基因融合检测结果比较 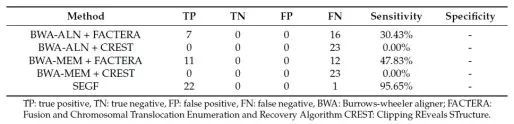 表 2 应用于临床样本上的基因融合检测结果比较  综上,由于大多数融合基因在基因之间都有一个模糊的断点,因此需要大量的计算时间去确定这些断点,元码研发的 SEGF 只保留双端 35bp 的准确序列进行比对,是一种不需要处理复杂参数的简单方法,但同时也保证了结果的准确性和灵敏度,为挖掘融合基因提供了一种新颖且高效的方法。 参考资料: 1.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. CANCER CELL 2002;2:117-125 2.https://www.cancer.gov/publications/dictionaries/cancer-terms/def/bcr-abl-fusion-gene 3.Li Y, Zheng H, Luo R, et al. Structural variation in two human genomes mapped at single-nucleotide resolution by whole genome de novo assembly. NAT BIOTECHNOL 2011;29:723-730 4.Shen R, Ying K, Wang Z, Schnable PS. ALGORITHM FOR DNA COPY NUMBER VARIATION DETECTION WITH READ DEPTH AND PARAMORPHISM INFORMATION Rong Shen1, Kai Ying2, Zhengdao Wang1, Patrick S. Schnable3. 2016 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP) 2016:869-873 5.Chen K, Wallis JW, McLellan MD, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. NAT METHODS 2009;6:677-681 6.Newman AM, Bratman SV, Stehr H, et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. BIOINFORMATICS 2014;30:3390-3393 7.Wang J, Mullighan CG, Easton J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. NAT METHODS 2011;8:652-654 来源: https://www.biomart.cn/news/10/2877923.htm
laihui126
2023年1月9日 15:06
分享文档
收藏文档
上一篇
下一篇
微信扫一扫
复制链接
手机扫一扫进行分享
复制链接
关于 MrDoc
觅道文档MrDoc
是
州的先生
开发并开源的在线文档系统,其适合作为个人和小型团队的云笔记、文档和知识库管理工具。
如果觅道文档给你或你的团队带来了帮助,欢迎对作者进行一些打赏捐助,这将有力支持作者持续投入精力更新和维护觅道文档,感谢你的捐助!
>>>捐助鸣谢列表
微信
支付宝
QQ
PayPal
下载Markdown文件
分享
链接
类型
密码
更新密码



